MRI SPINE (AXIAL)
A 4-year-old male child was brought by his parents with complaints of progressive back deformity (kyphosis) noticed since early infancy, and inability to walk. The parents reported that the child never achieved independent ambulation. There was no history of trauma. The mother denied any significant antenatal events, though no antenatal MRI or anomaly scan had been performed.
On further questioning, the parents noted that the child also had difficulty controlling bladder and bowel function. No similar illness was reported in any family member.
CHIEF COMPLAINT : 4 Years | Male
CHIEF COMPLAINT : Kyphosis + Inability to Walk
DURATION : Since Early Infancy
BLADDER/BOWEL : Incontinence Present
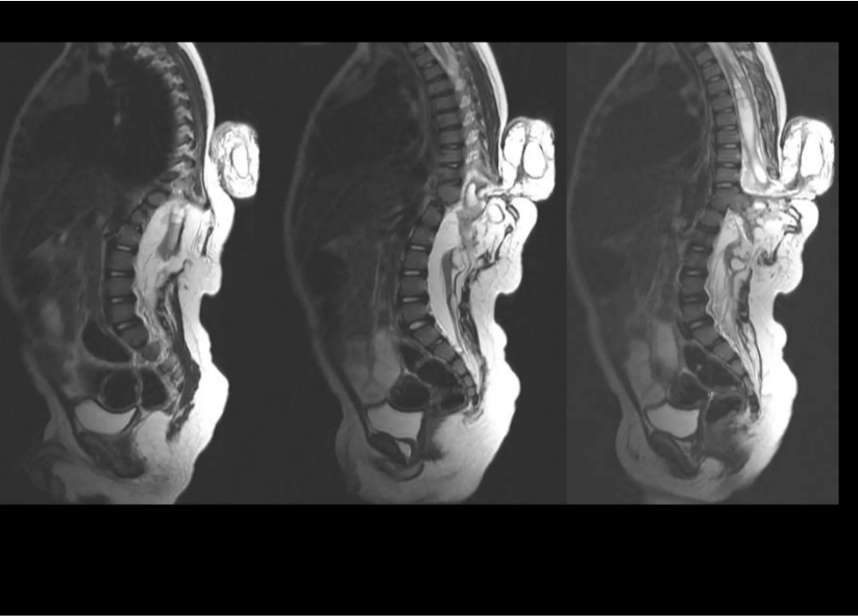
INVESTIGATION 1 — MRI SPINE (SAGITTAL)
T1 & T2 weighted sagittal sequences — Lumbar & Lumbosacral spine
- Severe thoracolumbar kyphosis
2. Large posterior lumbosacral spinal dysraphic defect
3. Fatty lipomatous mass within spinal canal
4. Low-lying tethered spinal cord
5. Syringohydromyelia — fluid-filled cord cavity
6. Posterior meningocele sac with cord tethering
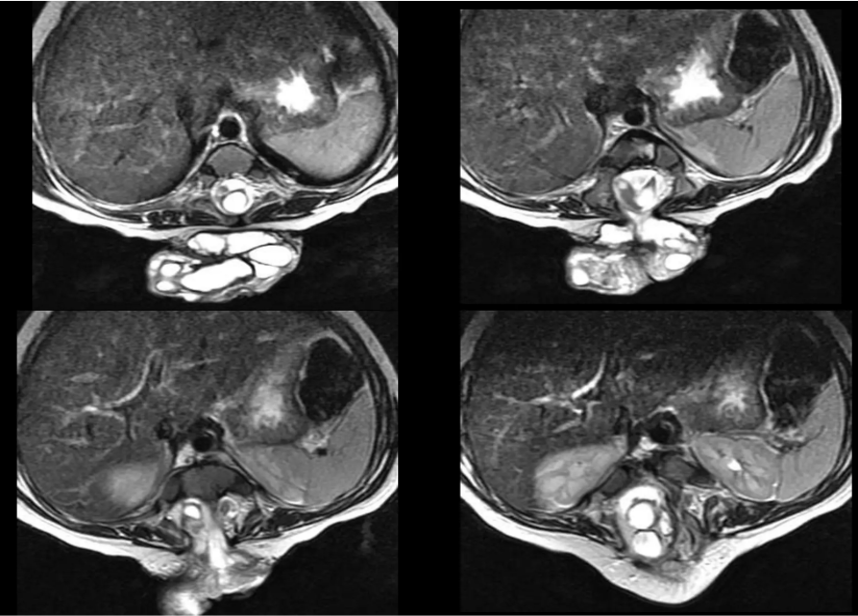
Findings
- Two separate hemicords visible — diagnostic of Diastematomyelia
2. Split cord malformation (Type I or II)
3. Each hemicord in its own dural sleeve
4. Bony or fibrous septum separating hemicords
5. Tethering of both hemicords confirmed
INVESTIGATION 2 — MRI SPINE (SAGITTAL)
T1 & T2 weighted sagittal sequences — Lumbar & Lumbosacral spine
Findings
1. Two separate hemicords visible — diagnostic of Diastematomyelia
2. Split cord malformation (Type I or II)
3. Each hemicord in its own dural sleeve
4. Bony or fibrous septum separating hemicords
5. Tethering of both hemicords confirmed
INVESTIGATION 2 — MRI BRAIN & CERVICAL SPINE
Sagittal & axial views — Craniocervical junction
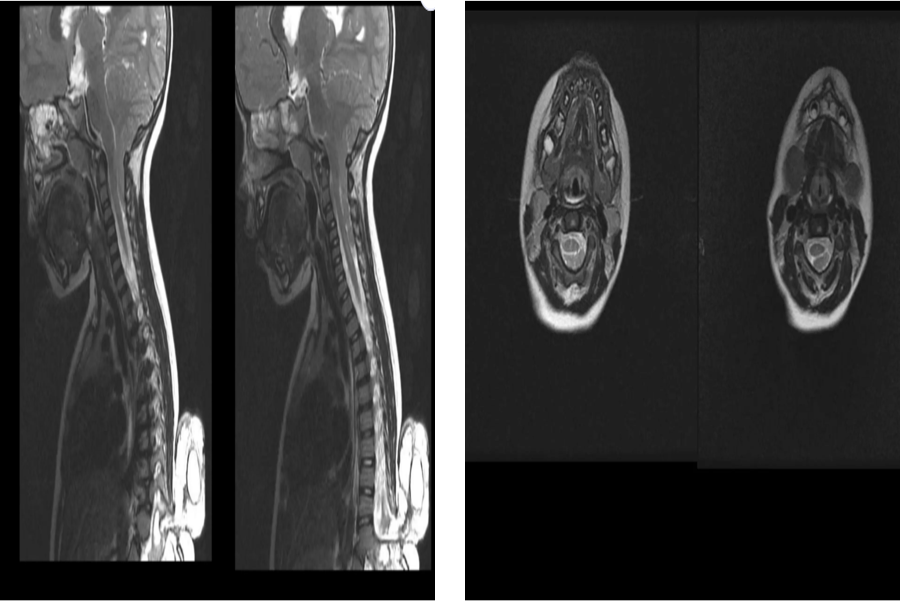
1. Cerebellar tonsillar herniation below foramen magnum
2. Arnold-Chiari Malformation Type I (ACM I)
3. Crowding at craniocervical junction
4. Cervical cord syrinx formation
5. Cervicomedullary compression
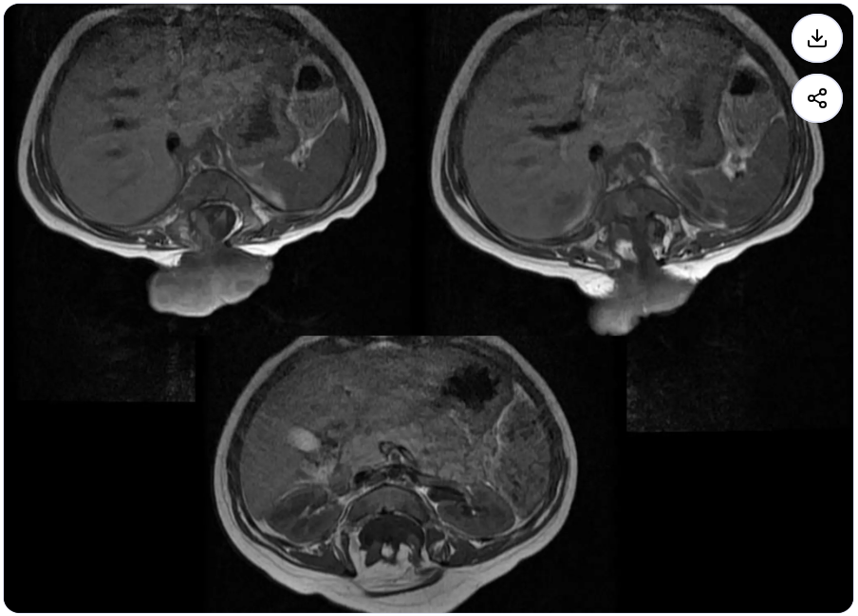
INVESTIGATION 3 — MRI ABDOMEN & PELVIS
Coronal sequences — extent of lipomeningomyelocele and pelvic involvement
1. Lipomeningomyelocele sac extending posteriorly
2. Large fatty presacral / pelvic mass
3. Intraspinal lipoma extending into spinal canal
4. Pelvic organ displacement by mass effect
5. Confirms multilevel dysraphic disease
FINAL DIAGNOSIS
- Lipomeningomyelocele sac extending posteriorly
2. Large fatty presacral / pelvic mass
3. Intraspinal lipoma extending into spinal canal
4. Pelvic organ displacement by mass effect
5. Confirms multilevel dysraphic disease
FINAL DIAGNOSIS
Based on Clinical Presentation + MRI Findings (Spine, Brain, Abdomen)

- Severe thoracolumbar kyphosis
2. Large posterior lumbosacral spinal dysraphic defect
3. Fatty lipomatous mass within spinal canal
4. Low-lying tethered spinal cord
5. Syringohydromyelia — fluid-filled cord cavity
6. Posterior meningocele sac with cord tethering
UNDERSTANDING THE DIAGNOSIS — LIPOMENINGOMYELOCELE
What is it?
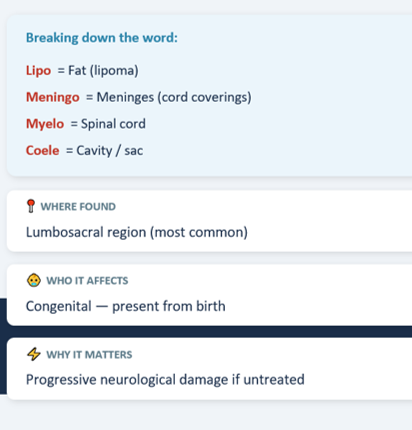
Lipomeningomyelocele is a form of occult spinal dysraphism where a fatty (lipomatous) mass is attached to the spinal cord and extends through a defect in the posterior spinal elements into the subcutaneous tissue.
The cord is tethered — held down — by this fatty mass, preventing it from ascending normally during growth Syringohydromyelia = a fluid-filled cavity within the spinal cord itself, caused by CSF flow obstruction due to tethering.
UNDERSTANDING THE DIAGNOSIS — DIASTEMATOMYELIA
What is it?
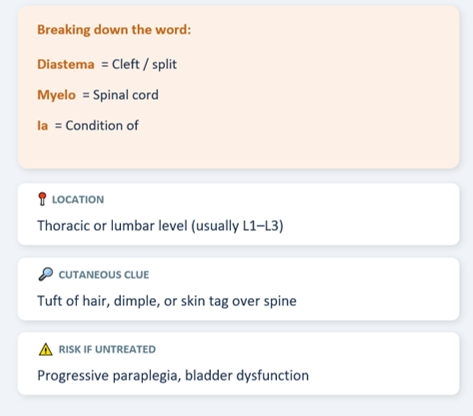
Diastematomyelia is a rare congenital anomaly where the spinal cord is split into two halves (hemicords) by a bony, cartilaginous, or fibrous septum.
Type I: Both hemicords in separate dural sleeves (more severe, more tethering).
Type II: Both hemicords share a single dural sac (less severe).
The split cords are tethered by the septum, causing progressive neurological deficit as the child grows and the spine lengthens.
UNDERSTANDING THE DIAGNOSIS — DIASTEMATOMYELIA
What is it?
Arnold-Chiari Malformation Type I (ACM I) is a structural defect where the lower part of the cerebellum (cerebellar tonsils) herniates downward through the foramen magnum into the spinal canal.
This causes crowding at the craniocervical junction and disrupts normal CSF flow, which can lead to syringomyelia (a fluid-filled cavity in the cervical cord).
Named after: Julius Arnold (German pathologist) and Hans Chiari (Austrian pathologist), who described the spectrum of hindbrain herniation malformations.
WHERE DO THESE CASES PRESENT?
Epidemiology, Origin & When to Suspect
Embryological Origin
All three conditions arise from defective primary or secondary neurulation — the process by which the neural tube closes between weeks 3–4 of fetal life. Failure leads to open or closed spinal dysraphism.
Population & Prevalence
Spinal dysraphism (all types): ~1 in 1000 live births globally. Higher in South Asia & Middle East. Lipomyelomeningocele alone: ~1 in 4000. Diastematomyelia: ~1 in 5000.
Where Patients Come From
Typically referred from pediatrics or general surgery after noting:
- Back swelling or skin lesion at birth
- Foot deformity (clubfoot, cavus)
- Asymmetric lower limb wasting
- Delayed walking or regression of milestones
- Recurrent UTIs / bladder dysfunction
What Raises Suspicion?
Always examine the lower back in any child with:
- Gait abnormality or toe-walking
- Asymmetric lower limb reflexes
- Unexplained bladder incontinence
- Visible lumbosacral swelling or hair patch
MANAGEMENT
What Can Be Done for This Patient?
Surgical Management
1. Cord Untethering — Primary surgery — release of tethered cord from lipomatous mass to prevent further neurological deterioration.
2. Lipoma Resection — Partial or total excision of the intraspinal lipoma; total excision risks cord damage.
3. Dural Reconstruction — Duraplasty — expansion of the dural sac to prevent re-tethering.
4. Foramen Magnum Decompression — For ACM I — posterior fossa craniectomy + C1 laminectomy to relieve cerebellar herniation.
5. Septum Resection — For diastematomyelia — removal of the bony/fibrous septum dividing the hemicords.
Non-Surgical Support
- Physiotherapy — strengthen lower limbs, gait training
- Orthotics / splints — foot drop management
- Clean intermittent catheterization — bladder care
- Bowel management program
Goals of Management
✓ Halt progression of neurological deficit
✓ Restore / improve bladder & bowel control
✓ Enable ambulation with or without aids
✓ Prevent re-tethering — long-term MRI follow-up
✓ Multidisciplinary care: neurosurgery + urology + physio
MANAGEMENT
What every clinician should remember about this case
1. Spinal dysraphism is a SPECTRUM — always do whole-spine MRI to look for associated anomalies (Chiari, syrinx, diastematomyelia).
2. A child with kyphosis + never walking + bladder issues = suspect closed spinal dysraphism until proven otherwise.
3. Surgery (untethering) should be done early and proactively — even before symptoms worsen — to preserve function.
4. ACM I is often clinically silent. Find it on MRI screen and monitor — decompression only if symptomatic or syrinx is expanding.